Research Supervisor
⚡ My research Supervisor is Dr. Shashank Tripathi, who is a Wellcome Trust India Alliance Intermediate Fellow in the Department of Microbiology & Cell Biology, Centre for Infectious Disease Research at Indian Institute of Science, Bangalore.

Research Interest
My research interest lies in exploring viral and bacterial infectious diseases, understanding the molecular mechanisms governing the disease establishment, host innate and adaptive immune response, as well as vaccine development and its efficacy testing. In this Ph.D. program, under the supervision of Dr. Shashank Tripathi, I seek serious specialization in the field of emerging viral diseases, viral biology, and antiviral innate immunity. My goal is to characterize the signaling pathway and the mediators of Interferon-independent antiviral innate immune response in mammalian cells. This study is expected to unravel a new class of anti-viral factors and mechanisms which will be tested against a wide range of human viruses including Coronaviruses, Influenza, Flaviviruses (Dengue, Zika), Retroviruses (HIV) and DNA viruses (Herpes). My long-term future objective is to study the extant as well as emerging viral pathogens and diseases, and to translate the knowledge acquired during doctoral and subsequent research into novel drugs and vaccination strategies for the betterment of the mankind..
Project 01
To study the role of IRF3 in Antiviral Innate Immune Response, it’s stability, interactions, negative regulation and mechanism of degradation.
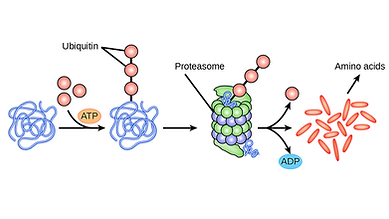
The general paradigm of mammalian antiviral response is the detection of viral pathogen-associated molecular patterns (PAMPs) by host Pattern Recognition Receptors (PRRs), which leads to activation of Interferon Regulatory Factors (IRFs). Activated IRFs (especially IRF3 and IRF7) turn on the transcription of Interferons (IFNs) which are critical mediators of the mammalian cellular antiviral innate immune response. IFNs act in an autocrine as well as paracrine fashion leading to activation of the JAK/STAT pathway which in turn activates the expression of a battery of antiviral genes, collectively called Interferon Stimulated Genes (ISGs). Although activated IRF3 is critical in the induction of IFN-β, a tight regulation of the IFN response and activated IRF3 is required within the cells. An unchecked and exaggerated expression of IFN-β can lead to severe immune-induced pathogenesis. Degradation of IRF3 is one of the mechanisms of negative regulation of IFN response. However, a mechanism of degradation and factors responsible are still minimally explored. Our study aims at determining the roles of IRF3 in IFN induction along with the background negative regulation of IFN response by the degradation of IRF3 to maintain immune homeostasis.
Project 02
Characterization of Broad Spectrum, Interferon signaling independent Antiviral Innate Immunity in Mammalian cells.
The signaling pathways leading to induction of IFN and subsequent expression of antiviral ISGs have been characterized in great detail. The elaborate sequence of events required for innate immune response to come in effect presents a window and enough time for the viruses to produce antagonists of host antiviral response, antagonize several signaling components and establish infection. It is plausible that mammalian cells may have alternate mechanisms to mount immediate antiviral response upon virus detection which is independent of elaborate IFN signaling and ISG induction. Indeed, we have discovered that a large subset of ISGs can be directly induced by IRFs in IFN signaling-deficient systems. Our study mainly focuses on identifying these antiviral factors and their mechanisms of action against a broad range of human viruses. The main objective of the project is to perform a detailed characterization of induction and action of antiviral factors which act in an IFN signaling-independent manner. This study will reveal new fundamental knowledge about mammalian antiviral immunity and facilitate the development of novel antiviral strategies.

PMRF Annual Reviews
Title: Characterization of Broad Spectrum, IFN Signaling-Independent Antiviral Innate Immunity in Mammalian Cells.
Title: Characterization of Broad Spectrum, IFN Signaling-Independent Antiviral Innate Immunity in Mammalian Cells
Introduction:
The general paradigm of mammalian antiviral response is the detection of viral pathogen-associated molecular patterns (PAMPs) by host Pattern Recognition Receptors (PRRs) which leads to the activation of Interferon Regulatory Factors (especially IRF3 and IRF7). IRFs turn on the transcription of Interferons (IFNs) which in turn activate the expression of a battery of antiviral genes, collectively called Interferon Stimulated Genes (ISGs). However, an unchecked and exaggerated expression of IFN-β can lead to severe immune-induced pathogenesis. Degradation of IRF3 is one of the mechanisms of negative regulation of IFN-β response. However, the mechanism of degradation and the factors responsible are still minimally explored. On the other hand, the elaborate sequence of events required for innate immune response to come into effect presents a window and enough time for the viruses to produce antagonists of the host antiviral response, antagonize several signaling components and establish infection. It is plausible that mammalian cells may have alternate mechanisms to mount an immediate antiviral response upon virus detection which is independent of elaborate IFN signaling and ISG induction. Indeed, we have discovered that a large subset of ISGs can be directly induced by IRFs in IFN signaling deficient systems. Our study mainly focuses on identifying these antiviral factors and their mechanisms of action against a broad range of human viruses. Our objectives are:
- To understand the regulation of IRF3 stability and function by cellular and viral factors
- To identify and characterize IRF3-dependent and IFN signaling-independent antiviral factors.
- To delineate IRF3 mediated transcription of antiviral genes in an IFN-independent manner.
Significance:
Successful execution of the proposed project will reveal new fundamental biology of the mammalian antiviral immune response. The new knowledge generated in this project will eventually facilitate the development of novel broad-spectrum antivirals and improved vaccination strategies in the future.
Work updates:
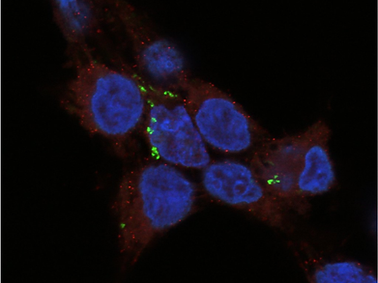
We showed that IRF3 is targeted both by proteasomal pathways and autophagosomes for degradation. This was done by studying the protein rescue upon treatment with proteasomal and autophagy inhibitors by western blotting (SDS-PAGE), Fluorescence-associated cell sorting (FACS), and Fluorescence microscopic analyses. We also found the triggering factors for IRF3 degradation. We also studied the effects of IRF3 rescue on IFN-β promoter activity by Renilla-Firefly dual-luciferase assay.

Review Results:
Overall Grade: Advanced to Next Scale
Remarks: Very well-designed work. Would recommend switching to more closer to endogenous models by using stable, low expression systems to study protein degradation.
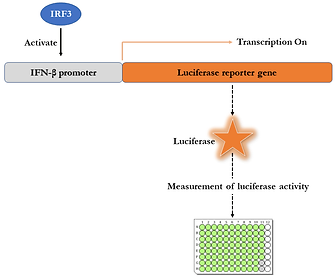
PMRF Annual review 2022:
Work updates:
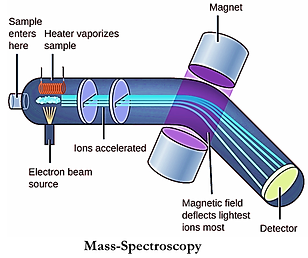
As recommended by the panelists in the previous review, we corroborated our findings at endogenous IRF3 protein levels by stimulating HEK-293T, and A549 cells with Poly I:C and/or SeV for checking the IRF3 and pIRF3 levels with and without drug treatments. We further moved on to dissect the mechanism of IRF3 degradation by studying the role of IRF3 ubiquitination/polyubiquitination and the roles of unique lysine residues on IRF3. Alanine scanning would determine the critical lysine residues. We are now in the process of alanine scanning of IRF3 protein, for which we have developed lysine mutants of IRF3-5D using PCR based site-directed mutagenesis. Also, the type of ubiquitin-linkage that play role in IRF3 stability and degradation was explored. IRF3 gene was cloned with 2X-C term strep tag and the proteins were purified using a Streptactin-purification kit for mass spectrometry-based analysis in order to determine the cellular interactors that may directly or indirectly contribute to its stability within the cells. Mass spectrometry was performed using the Orbitrap fusion MS facility and the results are currently under analysis. On the other hand, for the CRISPR screening of host factors regulating IRF3-WT and IRF3-5D degradation, a genome-wide CRISPR knock-out library of HEK-293T cells has been generated by transducing them with CRISPR lentiviruses followed by selection for 7 days. These cells have been transfected (24hrs) with IRF3-GFP (biological replicates n=3) and the high GFP population has been sorted in the BD-Aria-II sorter facility. The genomic DNA from these sorted cells will be isolated, PCR amplified, and sent for sequencing for further analysis which will reveal the essential host factors underlying IRF3 degradation.
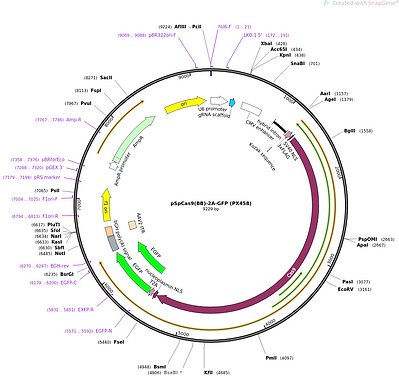
We have successfully knocked out STAT1 in A549 and HEK-293T and has been validated by immunoblotting for endogenous STAT1. The STAT1-/- A549 and HEK293T cells were treated with IFN for 12 hrs and then harvested to carry out RT-PCR for a few ISGs (ISG54, ISG56, and 18S rRNA control). This further helped corroborate the successful knockout of STAT1 from these cell lines. We have also cloned gDNA against STAT2, IRF7, IRF9, and IRF1 in the pSP-cas9-2A-GFP backbone in order to create single and double knockouts in A549 and HEK-293T cells which will be used later to delineate IRF3-mediated transcription of antiviral genes in IFN-independent manner. We are currently in the process of making the lentiviral constructs of the genes that are upregulated in an IRF3-dependent but IFN-independent manner. For testing the antiviral potential of these genes, a panel of reporter viruses will be used. We have the stocks of reported viruses prepared and titrated.
Review Results:
Review Comment May 2022: Recommended with commendation
Detailed Review Comment May 2022:
Very good progress. Future plans are well defined. Validation in STAT1 and STAT2 knockout may help validate findings.